
Remember me
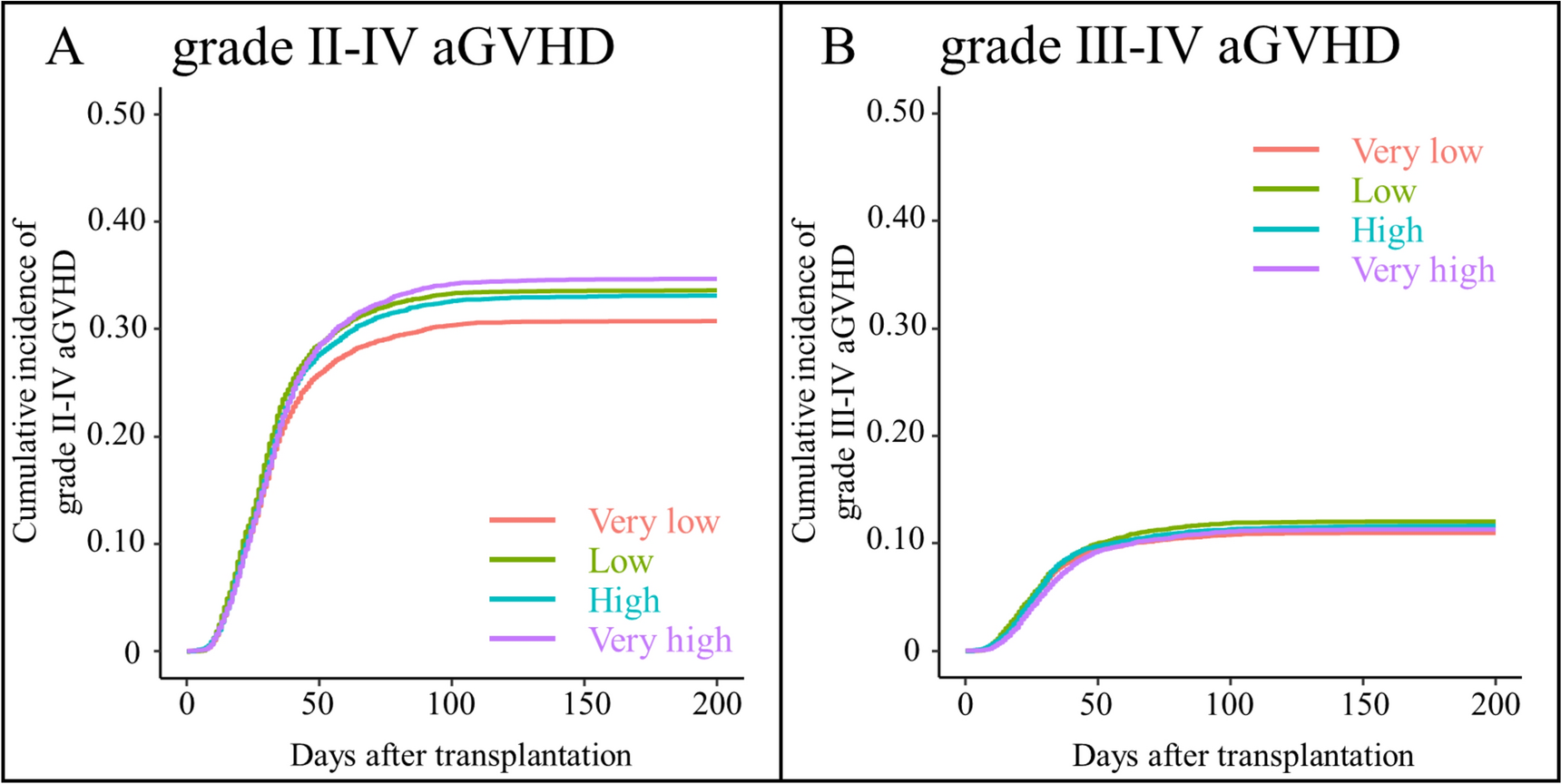
Overall, 273 patients were registered for the PMS, and eCRFs were collected from 259. The safety analysis set included 248 patients and the effectiveness analysis set included 202 patients (Fig. 1).
Fig. 1
Post-marketing surveillance populations
Patients in the safety analysis set were aged 26.0–91.0 (median 74.0) years (Table 1), with most (69.4%) aged ≥ 70.0 years. The majority of patients were male (77.8%). The indication for ibrutinib was refractory MCL in 73 patients (29.4%) and relapsed MCL in 175 (70.6%), and most patients (n = 173 [69.8%]) had Ann Arbor stage IV disease. In total, 117 patients (47.2%) had t(11; 14) translocation, 185 (74.6%) had expression of cyclin D1 identified, and 31 (12.5%) had bulky disease. MCL was confirmed by genetic testing in 199 patients (80.2%). Patients had received a median (range) of 3.0 (1.0–12.0) prior lines of therapy. The most common prior therapy was combination therapy with rituximab + bendamustine (Supplementary Table S1, Electronic Supplementary Materials).
Table 1 Demographic and baseline clinical characteristics (safety analysis set)aIbrutinib treatmentThe median (range) follow-up duration was 6.1 (0.1–13.1) months. At the end of follow-up, 98 patients were still receiving ibrutinib and 150 (60.5%) had discontinued treatment, mostly because of insufficient effect (n = 56 [22.6%]), AEs (n = 39 [15.7%]), or death (n = 28 [11.3%]; Table 2). Eighty-seven patients (35.1%) required a dose reduction or a temporary interruption, mainly because of AEs (n = 78 [31.5%]; Table 2).
Table 2 Treatment details and patient disposition (safety analysis set)The mean ± SD daily dose of ibrutinib during the observation period was 456.2 ± 129.5 mg.
EffectivenessIn the effectiveness analysis set (n = 202), the ORR was 59.9%, and the median time to best response was 77.0 days. Fifty-six patients (27.7%) achieved CR, 65 (32.2%) had PR, 34 (16.8%) had stable disease, and 46 (22.8%) had PD. Data from one patient in the effectiveness analysis set was classified as “missing/unlisted", even though a best response was available for this patient (PR). For this patient, the end date of the observation period was incorrectly entered and, as such, we were unable to determine if this effect measurement was performed within the observation period. A total of 94 patients had PD at some point during the observation period (regardless of final assessed ORR); of these, 53 patients continued to receive ibrutinib for a duration of ≥ 2 days (median 26 days) after diagnosis of PD.
The 52-week PFS rate was 47.5%, and the 52-week OS rate was 69.3%. Median PFS was 320.0 days (95% confidence interval: 208.0 days, not reached; Fig. 2a) and the median OS had not been reached (Fig. 2b).
Fig. 2
a Progression-free survival and b overall survival during ibrutinib treatment. Data are for the effectiveness analysis set. No. number, NR not reached, OS overall survival, PFS progression-free survival
SafetyOverall, 74.6% of patients developed an AE. The most common AEs occurring in ≥ 3.0% of patients were platelet count decreased (occurring in 10.4% of patients), pneumonia (7.2%), neutrophil count decreased (6.8%), diarrhea (6.0%), anemia (6.0%), decreased appetite (4.4%), white blood cell count decreased (4.0%), and malaise (3.2%; Fig. 3).
Fig. 3
Adverse events (other than mantle cell lymphoma progression and death) occurring in ≥ 3% of patients. WBC white blood cell
Approximately 67.0% of patients developed an AE not related to MCL progression events. Of these AEs, approximately 50.0% were Grade 1 or 2 in severity, and 50.0% were Grade ≥ 3 (Fig. 4). The incidence of these AEs was slightly higher in patients who had received one previous line of therapy than in those who had received > 1 previous line of therapy, and in those aged ≥ 70.0 years than those < 70.0 years (Fig. 4).
Fig. 4
Adverse events (other than mantle cell lymphoma progression and death) by grade, overall and in patient subgroups. PLOT previous lines of therapy
AEs of special interestInfections, bleeding, and arrhythmias of any grade occurred in 19.8%, 10.1%, and 2.0% of patients, respectively, and were of Grade ≥ 3 severity in 9.7%, 2.4%, and 0.4% of patients, respectively (Table 3).
Table 3 Dose management and outcomes of adverse events of special interest (safety analysis set)Infections that occurred in ≥ 5 patients were pneumonia (n = 17, 6.9%), bronchitis (n = 6, 2.4%), and herpes zoster (n = 5, 2.0%; Supplementary Table S2, Electronic Supplementary Materials). Three patients experienced Grade 5 infections that were attributed to ibrutinib treatment. Of these, one patient, who also had Grade 3 anemia, developed bacterial sepsis and pneumonia after approximately 20 weeks of ibrutinib treatment; ibrutinib was immediately discontinued, but the patient died 17 days later. One patient developed meningitis approximately 9 weeks after stopping ibrutinib, having received the drug for approximately 20 weeks prior; they also had Grade 1 pneumonia, Grade 3 herpes zoster oticus, and Grade 3 platelet count decreased. The patient died 5 days after developing meningitis. The third patient developed bacterial pneumonia after approximately 4 weeks of ibrutinib treatment; ibrutinib was discontinued, but the patient died 13 days later.
Overall, 120 (48.3%) patients in the safety analysis set received anti-infective prophylaxis and 30 developed infections (25.0%), compared with 20 infections in the 128 patients who did not receive prophylaxis (15.6%; Supplementary Tables S3 and S4, Electronic Supplementary Materials). The most common prophylaxis was drugs used to treat Pneumocystis jirovecii pneumonia (trimethoprim/sulfamethoxazole or atovaquone). Two of the three infection-related deaths occurred in patients who had received anti-infective prophylaxis.
The number of patients requiring dose reduction, interruption, or ibrutinib discontinuation due to AEs of special interest is shown in Table 3. Overall, infections were the cause of permanent treatment discontinuation in seven patients (all Grade ≥ 3), an ibrutinib dose reduction in six (Grade 1 or 2 in five and Grade ≥ 3 in one), and a temporary dose interruption in 25 (Grade 1 or 2 in 14 and Grade ≥ 3 in 11). However, five of the six patients who underwent a dose reduction were able to re-escalate the ibrutinib dose again, and 22 of the 25 for whom a temporary dose interruption was put in place were able to restart ibrutinib after the interruption. Infections completely resolved in 87.8% of patients (43/49 of those with infection) who developed an infectious episode of any grade.
The only bleeding event that occurred in ≥ 5 patients was petechiae (n = 5, 2.0%; Supplementary Table S2, Electronic Supplementary Materials). Anticoagulant or antiplatelet agents were being used by 48 patients in the safety analysis set (19.4%), and nine of these patients developed bleeding events (18.8%); the other 16 bleeding events occurred in patients not taking anticoagulant or antiplatelet agents (8.0%). The type and doses of anticoagulant/antiplatelet therapies are shown in Supplementary Table S5 in the Electronic Supplementary Materials. Five of the 16 patients taking aspirin and four of the six patients taking a direct-acting oral anticoagulant (DOAC) developed bleeding during ibrutinib treatment.
Bleeding led to permanent ibrutinib discontinuation in six patients (two with Grade 1 or 2 and four with Grade ≥ 3 bleeding), an ibrutinib dose reduction in five (all Grade 1 or 2), and a temporary dose interruption in eight (six with Grade 1 or 2 and two with Grade ≥ 3 bleeding; Table 3), but two of the five patients were able to increase the ibrutinib dose again, and all eight were able to restart ibrutinib after the temporary interruption. Bleeding completely resolved in 80.0% of patients who developed a bleeding episode of any grade.
The five patients who experienced arrhythmias all developed atrial fibrillation (AF; Supplementary Table S2, Electronic Supplementary Materials); AF developed in two of 13 patients who had concurrent arrhythmia at baseline (15.4%) and in three of 230 patients with no concurrent arrhythmias (1.3%; Supplementary Table S6, Electronic Supplementary Materials). No patients with a past medical history of arrhythmia developed AF during ibrutinib treatment. Three patients who developed AF (two with Grade 1 or 2 AF and one with Grade ≥ 3 AF) permanently discontinued ibrutinib (Table 3). Another patient who developed AF had a temporary dose interruption, but was able to restart ibrutinib again once the AE had resolved. The remaining patient who developed AF did not require a dose change. AF completely resolved in three patients (60.0%) who developed AF of any grade.
The incidence of AEs of special interest, including Grade ≥ 3 AEs, was higher during the first 10 weeks after ibrutinib initiation, and decreased thereafter (Fig. 5).
Fig. 5
Timing of onset for adverse events of special interest of a–e all grades and f–j Grade ≥ 3 in the safety analysis set (N = 248)
Comments (0)