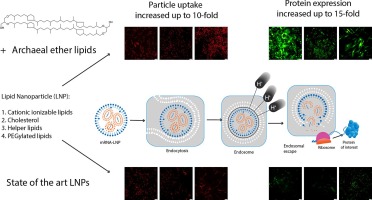
Over the last decades, lipid nanoparticles (LNPs) have evolved to become the most advanced nonviral platform for safe and highly effective nucleic acid delivery in vivo, since they can encapsulate and deliver a wide variety of therapeutic agents. The first FDA approved small-interfering RNA (siRNA) drug, Onpattro (Patisiran), is an LNP formulation that after intravenous application delivers siRNA into liver hepatocytes for the treatment of hereditary transthyretin amyloidosis [1]. More recently, messenger RNA (mRNA) LNPs have gained importance as intramuscularly applied vaccines in the context of the COVID-19 pandemic [2] and for other vaccination directions, such as antitumoral immunotherapy [3]. In addition, other work uses LNPs for the co-delivery of different nucleic acid molecules, such as CRISPR-Cas9 mRNA and single guide RNA to the liver, as evaluated in a recent clinical study for the treatment of transthyretin amyloidosis [4].
LNPs typically consist of four lipidic components: cholesterol, phospholipids, polyethylene glycol (PEG)-lipids, and cationizable lipids [5]. These components enable monodisperse particle formation and high encapsulation efficiency of nucleic acid, improve particle stability, and promote endosomal escape of nucleic acid after cellular uptake via endocytosis. The main focus in LNP research is the optimization of ionizable lipids since these are mainly responsible for efficient complexation of the negatively charged nucleic acid and endosomal escape after cellular uptake. Most recently, LNP optimization through variation of phospholipids, PEG lipids, replacement of cholesterol as well as ratio optimization of the individual components has come to the fore [6], [7]. For example, by replacing zwitterionic 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) with anionic 1,2-distearoyl-sn-glycero-3-phosphoglycerol (DSPG) in Onpattro (Patisiran), the LNP surface charge can be switched from neutral to anionic and thus target the reticuloendothelial system (RES) [8]. Furthermore, the combination of appropriate helper lipids is crucial. By changing the head group of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) to ethanolamine (DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), the LNP formulation containing cationic lipid DODAP (1,2-dioleoyl-3-dimethylammonium propane) facilitates a transition from the LDL (low density lipoprotein) receptor-mediated hepatocyte delivery via apolipoprotein E (ApoE) to predominant spleen targeting via complement receptors [9], [10]. Currently, the most advanced LNP formulations base on standard cationizable lipids with a single positive charge or lipidoids with several positive charges [11], [12]. In vivo, the LNP composition dictates protein corona formation in the bloodstream, which in turn affects biodistribution and cellular uptake. Due to ApoE-mediated liver targeting and good blood perfusion, the liver represents an ideal organ for LNP delivery and a major site of drug metabolism [13]. In addition to that, the LNP composition influences the physico-chemical properties (particle size, charge, surface shielding), which enables organ-selective targeting in the absence of receptor targeting moieties. For example, particle size can be regulated by altering the PEG lipid concentration to reach hepatocytes by passing through liver fenestrae (100–160 nm depending on species) [14]. Further, negatively charged particles show high spleen preference as reported for lipoplexes [15]; and larger particle sizes, ranging from 200 to 500 nm, are beneficial for targeting splenic dendritic cells [16].
Despite the potent delivery of LNPs to the liver and the overall established medical usefulness of hepatocyte-directed nucleic acid therapies, it may be desirable to redirect the particles to other therapeutically relevant tissues. A new approach to minimize liver accumulation is selective organ targeting (SORT). This method involves the addition of a fifth, “sorting” molecule to the lipid composition that determines the tissue-specific delivery and activity of the LNPs [17], [18], [19]. Most recently, peptide ligands specifically binding receptors/cell types were identified by in vivo phage peptide biopanning and conjugated to mRNA LNPs for photoreceptor targeting of the neutral retina after sub-retinal injection [20]. Additionally, there are strategies to address leukocytes; e.g., conjugation of a fusion protein, which addresses α4β7 integrin expressed on gut-homing leukocytes, to the surface of the LNPs resulted in interferon γ silencing in the gut and therapeutic efficacy in experimental colitis [21]. Another approach is the design of new libraries of ionizable amino lipids based on ethanolamine, hydrazine, or hydroxylamine linkers for efficient siRNA delivery into leukocytes [22]. Library screening and evaluation with Fast Identification of Nanoparticle Delivery (FIND), a high-throughput DNA barcode-based in vivo LNP screening, facilitates the characterization of chemically distinct LNPs for efficient RNA delivery to T-lymphocytes or splenic immune cells in vivo via chemical targeting [23], [24]. Also, the additional incorporation of the bioactive phospholipid phosphatidylserine into LNPs promotes mRNA transfection efficiency in secondary lymphoid organs both in vitro and in vivo [25], [26].
A critical issue in successful nucleic acid delivery is the endosomal escape. Due to their ionizable character, LNPs are neutral at physiological pH, but become protonated in the acidic environment of endosomes. This aids in the fusion of their lipids with the endosomal membrane and facilitates nucleic acid delivery to the cytosol [27], [28]. Nevertheless, most of the cargo remains inactive by accumulation in late endosomes and lysosomes [5], [29]. There are several factors and limitations to endosomal escape that can hinder the efficiency of nanocarrier-mediated drug delivery, resulting in a cargo escape rate of only 1–2% [30], [31]. Overcoming these barriers is critical for the efficiency of therapeutic agents [32], [33].
The present study aimed at the evaluation of mRNA LNP formulations with improved intracellular cargo release. For this purpose, recently developed double pH-responsive lipo-xenopeptides [34] were evaluated as cationizable components in mRNA LNPs.
These sequence-defined xenopeptides were synthesized via solid phase-assisted peptide synthesis (SPPS). The double pH-responsiveness was implemented by combining cationizable polar aminoethylene units in form of the artificial amino acid succinoyl tetraethylene pentamine (Stp) [35], with cationizable apolar units in form of lipo amino fatty acids (LAFs) [34], yielding different 2D sequences and 3D topologies. These LAF-Stp carriers show a molecular chameleon character due to the pH-dependent switch in polarity and high endosomolytic activity. Selected library members were already found highly effective in standard mRNA lipopolyplexes [34]. To identify the best suitable LAF-Stp carriers for mRNA LNPs in comparison to standard ionizable lipids that contain only single tertiary amines (i.e., SM-102, and DLin-MC3-DMA) [2], [5], [36], [37], our multi-protonatable carriers with 6–10 protonatable amines were tested in different molar lipid, molar charge, and N/P ratios.
The LAF-Stp carriers formulated as LNPs turned out to be very potent, mediating high mRNA expression in vitro and in vivo, with an interesting specificity for the spleen as a central immune organ, displaying a far higher spleen/liver expression ratio than established standard SM-102 and DLin-MC3-DMA LNPs. All in all, our novel LAF-Stp carriers proved to be an interesting alternative to ionizable lipids in LNPs.



Comments (0)